
Cell-type-resolved genetic variation shapes inflammatory bowel disease risk
Most genetic variants associated with complex diseases lie in non-coding regions, complicating efforts to identify effector genes and relevant cell types.
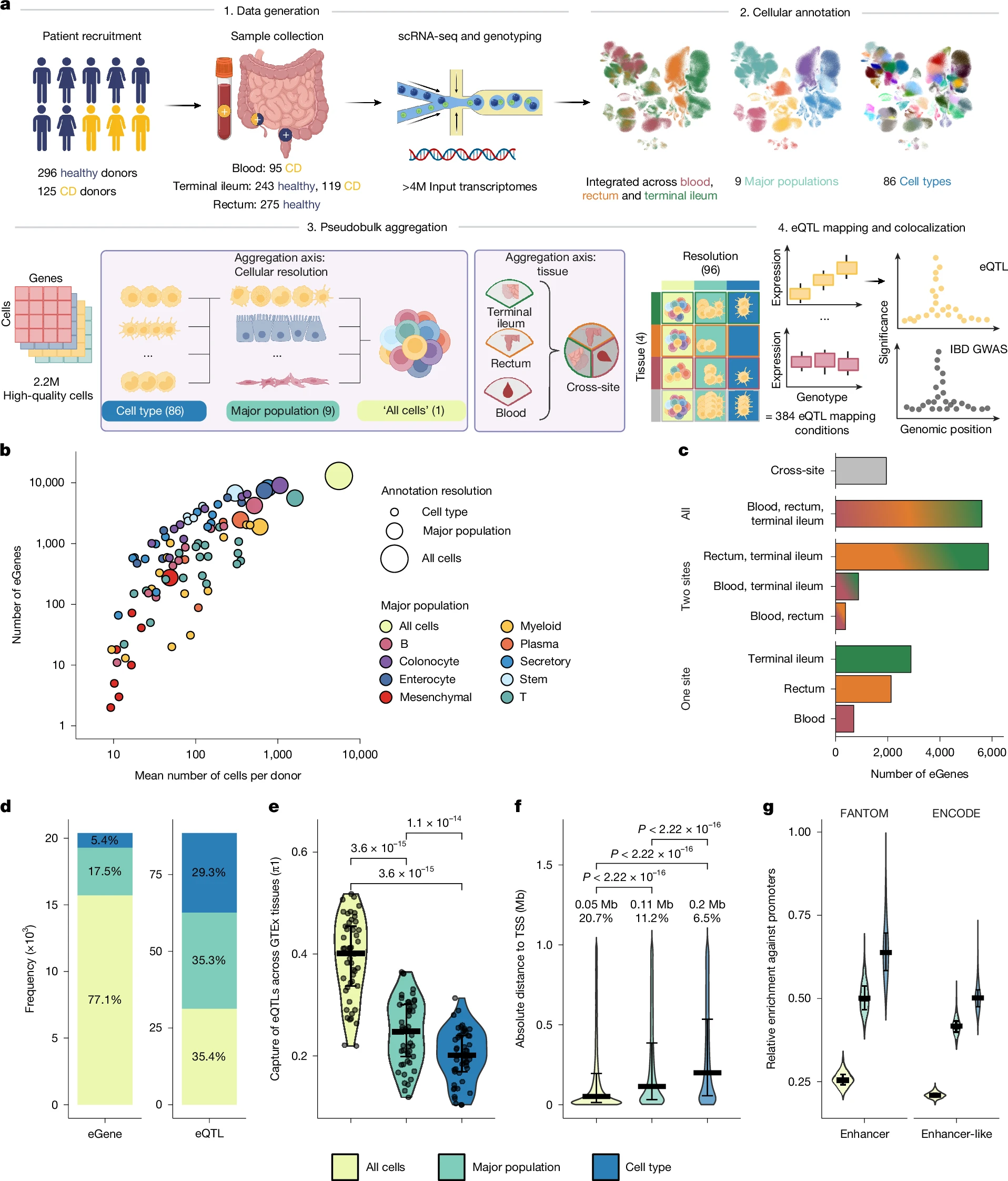
Here we map cis-expression quantitative trait loci (eQTLs) across 2.2 million single cells using intestinal biopsies and blood from 421 individuals, including 125 with inflammatory bowel disease (IBD). Cell-type-level eQTLs were more distal to transcription start sites, enriched in enhancers, less likely to regulate the nearest gene, and more than 3.5-fold more likely to colocalize with IBD loci detected in genome-wide association studies (GWASs) than eQTLs detected at tissue-level resolution.
We nominate effector genes at more than half of known IBD loci, including MAML2, PSEN2 and ZMIZ1 in myeloid cells, implicating reduced Notch signalling in intestinal immune dysfunction. We also identify Wnt-regulated genes, including MYC, in epithelial stem and progenitor cells, suggesting that impaired renewal contributes to barrier breakdown. Our results provide a mechanistic map that links genetic risk to specific genes and cell types in IBD, and a generalized framework for interpretation of GWAS loci using single-cell eQTL mapping of disease-relevant tissues in complex diseases.
Reference: Tobi Alegbe et al, Nature ,2026
Genomic analyses implicate hormonal and metabolic dysregulation in polycystic ovary syndrome
Polycystic ovary syndrome (PCOS) and its underlying features remain poorly understood. In this genetic study (n = 544,513), we expand the number of genetic loci from 16 to 29, and additionally identify 31 associated plasma proteins.

Many risk-increasing loci were associated with later age at menopause, underscoring the reproductive longevity related to an increased oocyte number and/or availability across the lifespan. Hormonal regulation in the etiology of this condition, through metabolic and reproductive features, was emphasized. The proteomic analysis highlighted metabolic biology known to be related to PCOS. A polygenic risk score (PRS) was associated with adverse cardiometabolic outcomes, with differing relevance of testosterone and body mass index in women and men.
Finally, while oligo-anovulation and anovulatory infertility are features of PCOS, we observed no impact of PCOS susceptibility on childlessness. We suggest that PCOS susceptibility confers balanced pleiotropic influences on fertility in women, and life-long adverse metabolic consequences in both sexes.
Reference: Loes M. E. Moolhuijsen et al, Nature Genetics 2026
The exposome of brain aging across 34 countries
The physical and social exposome affects human aging, and brain clocks may track its effects. However, most studies neglect multidomain exposures (physical, social and political) across diverse settings globally and their associations with brain aging.
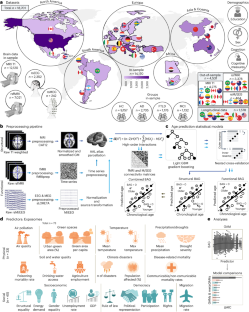
In this study, we characterized the associations between 73 country-level physical and social exposomal factors and multimodal brain age in 18,701 participants from 34 countries (healthy individuals and those with Alzheimer’s disease, frontotemporal lobar degeneration or mild cognitive impairment). Exposome effects were assessed using generalized additive models and meta-analytic frameworks. Aggregated exposome models explained up to 15.5-fold more variance than individual exposures (delta Akaike information criterion (ΔAIC): 2,034–3,127).
Physical exposome was primarily associated with accelerated structural brain aging (limbic, subcortical and cerebellar regions), whereas social exposome was more strongly associated with functional brain aging (frontotemporal and limbic networks). Exposome burden accounted for 3.3−9.1-fold higher risk of accelerated aging, exceeding effects of clinical diagnoses. Findings were out-of-sample validated in cross-sectional and longitudinal designs, remained consistent across clinical subgroups and persisted after adjustment for demographics, age correction bias, cognition, scanner type and data quality. The exposome accelerates brain aging in health and disease, underscoring the need to address physical, social and political inequities.
Reference: Agustina Legaz et al Nature Medicine, 2026
Single-cell spatial transcriptomic analysis of human skin anatomy
The skin is the largest human organ and a site of substantial disease burden, yet its cellular and molecular organization across the body is largely undefined.

Here we construct an organ-wide single-cell spatial atlas of ~1.2 million cells from normal adult human skin, resolving the location of 45 cell types across 114 samples encompassing 15 anatomic sites. We uncover site-specific stereotypic cell-type composition and their organization into ten multicellular neighborhoods, most notably a perivascular neighborhood reminiscent of skin-associated lymphoid tissue. Within this neighborhood, ligand–receptor (L–R) analyses identify a central role for tumor necrosis factor in maintaining CCL19+ perivascular fibroblasts, highlighting homeostatic immune–stromal crosstalk.
Finally, comparing neighborhood dynamics in spatial transcriptomics of skin disease, we find pan-disease immune alterations in this perivascular neighborhood, suggesting spatial compartmentalization of pathogenic activity. Thus, multicellular neighborhoods underlie the skin’s multiscale molecular to macroanatomic organization, orchestrate cell–cell interactions and anatomic site specialization and exhibit architectural disruption in disease.
Reference: Paula Restrepo et al, Nature 2026
Immune-microbiome coordination defines interferon setpoints in healthy humans
Human immune systems are highly variable, with most variation attributable to non-genetic sources. The gut microbiome crucially shapes the immune system; however, its relationship with the baseline immune states of healthy humans remains incompletely understood.

Therefore, we performed multi-omic profiling of 110 healthy participants through the ImmunoMicrobiome study. A factor-based integrative approach identified coordinated variation, revealing that the interferon response was amongst the most variable immune features in healthy participants.
Microbiome composition, pathways, and stool metabolites varied concomitantly with interferon response pathways. Longitudinal data spanning more than a year indicated the significant stability of these parameters within individuals over time. Our study provides extensive data to examine the relationship between the immune states and microbiomes of healthy individuals at steady state, which paves the way for delineating inter-individual differences relevant for disease susceptibility and responses to therapy.
Reference: Joel Babdor et al , Cell, 2026
Individualized mRNA vaccines evoke durable T cell immunity in adjuvant TNBC
Triple-negative breast cancer (TNBC) is frequently associated with metastatic relapse, even at an early stage.
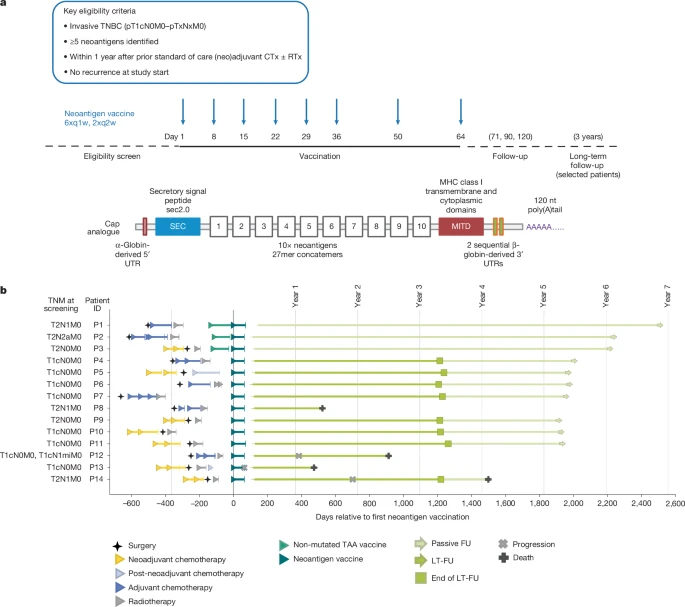
Here we assessed an individualized neoantigen mRNA vaccine in 14 patients with TNBC following surgery and after neoadjuvant or adjuvant therapy. In peripheral blood of nearly all patients, high-magnitude, vaccine-induced, mostly de novo T cell responses to multiple neoantigens were detected that remained functional for several years. Characterization of individual patients revealed that a large proportion of these T cells developed into two subsets: a late-differentiated phenotype with markers indicative of ‘ready-to-act’ cytotoxic effector T cells, and T cells with a stem cell-like memory phenotype. Eleven patients remained relapse-free for up to six years post-vaccination.
Recurrence occurred in three patients: the individual with the weakest vaccine-induced T cell response relapsed, but achieved complete remission on subsequent anti-PD-1 therapy; another patient had a tumour with low major histocompatibility complex (MHC) class I expression with MHC class I-deficient cells growing out under vaccination; and the third patient was BRCA-positive and had a recurrence from a genetically distinct primary tumour. These findings demonstrate the feasibility of individualized RNA vaccines in TNBC, document persistence of vaccine-induced, functional neoantigen-specific T cells and provide insights into possible immune escape mechanisms that will guide future approaches.
Reference: U. Sahin et al, Nature 2026
GWAS of major anxiety disorders in 122,341 European-ancestry cases
The major anxiety disorders (ANX; including generalized anxiety disorder, panic disorder and phobias) are highly prevalent, often onset early and cause substantial global disability.
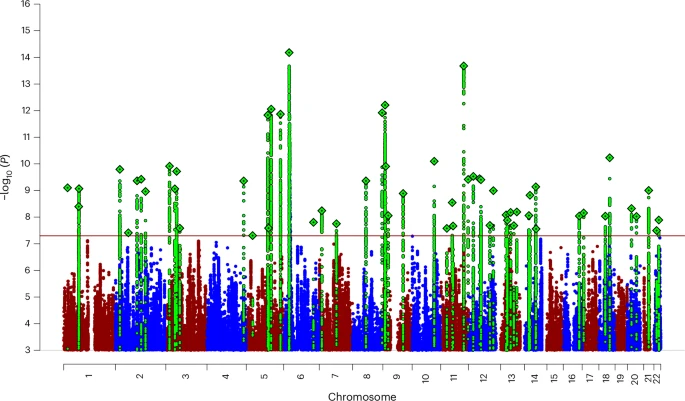
Although distinct in their clinical presentations, they probably represent differential expressions of a dysregulated threat–response system. Here, we present a genome-wide association meta-analysis comprising 122,341 European ancestry ANX cases and 729,881 controls. We identified 58 independent genome-wide significant risk variants and 66 genes with robust biological support. In an independent sample of 1,175,012 self-report ANX cases and 1,956,379 controls, 51 out of the 58 associations replicated.
As predicted by twin studies, we found substantial genetic correlation between ANX and depression, neuroticism and other internalizing phenotypes. Follow-up analyses demonstrated enrichment in all major brain regions and highlighted GABAergic signaling as one potential mechanism implicated in ANX genetic risk. These results advance our understanding of the genetic architecture of ANX and prioritize genes for functional follow-up studies.
Reference: Nora I. Strom et al, Nature genetics , 2026
Integrative epigenetics and transcriptomics identify aging genes in human blood
Recent epigenome-wide studies have identified a large number of genomic regions that consistently exhibit changes in their methylation status with aging across diverse populations, but the functional consequences of these changes are largely unknown.
On the other hand, transcriptomic changes are more easily interpreted than epigenetic alterations, but previously identified age-related gene expression changes have shown limited replicability across populations. Here, we develop an approach that leverages high-resolution multi-omic data for an integrative analysis of epigenetic and transcriptomic age-related changes and identify genomic regions associated with both epigenetic and transcriptomic age-dependent changes in blood.
Our results show that these multi-omic aging genes in blood are enriched for adaptive immune functions, replicate more robustly across diverse populations and are more strongly associated with aging-related outcomes compared to the genes identified using epigenetic or transcriptomic data alone. These multi-omic aging genes may serve as targets for epigenetic editing to facilitate cellular rejuvenation.
Reference: Mahdi Moqri et al Nature communications, 2026
zAvatar-test- A functional precision model to personalize ovarian cancer treatments: Results from a co-clinical study
In ovarian cancer, 80% of patients relapse after first-line therapy. In recurrent cases, oncologists lack reliable tests to guide chemotherapy choices, creating an unmet clinical need.

Here, we develop the ovarian cancer zebrafish Avatar-test, a functional in vivo model using patient tumor cells implanted in zebrafish embryos to predict treatment responses. We present the largest observational study (32 patients), where the zAvatar-test achieves 91% accuracy in predicting patient outcomes. Patients with a zAvatar-sensitive-test correlate with longer progression-free survival (17 vs. 6 months).
Tumors in zAvatars are dynamic, with human-host cell interactions, and higher metastatic potential in poor-prognosis cases. Finally, as a proof of concept, we demonstrate that venetoclax has the potential to sensitize multidrug-resistant tumors. Altogether, this clinical study demonstrates that the zAvatar-test may help clinicians personalize treatments for ovarian cancer patients. We are now conducting a multicentric randomized clinical trial to evaluate the zAvatar-test as a companion tool in clinical oncology.
Reference: Marta F. Estrada et al, Cell Reports Medicine, 2025
Genomic and transcriptomic analyses of aortic stenosis enhance therapeutic target discovery and disease prediction
Aortic stenosis (AS) is a common valvular heart disease and has no pharmacological therapies.
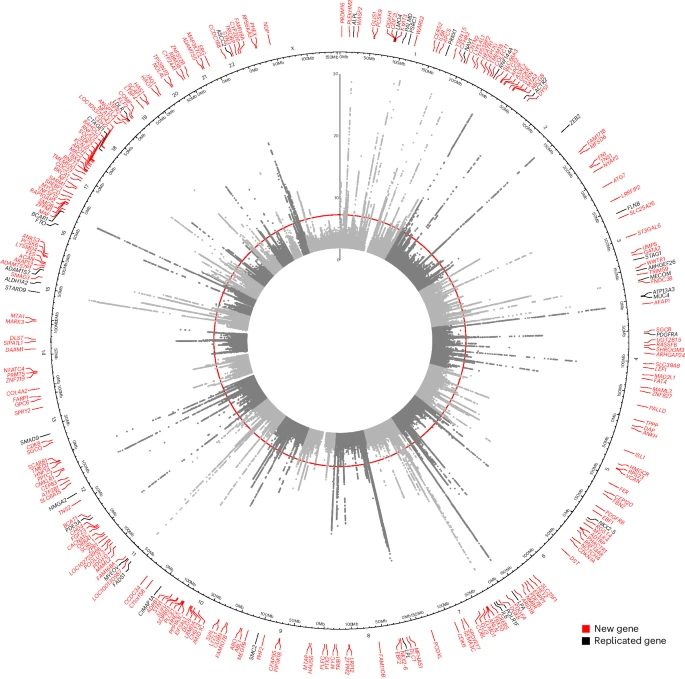
We performed a multi-ancestry genome-wide association meta-analysis of 86,864 AS cases among 2,853,408 individuals, discovering 241 autosomal independent risk loci and 3 X chromosome risk loci. We additionally performed sex-stratified and ancestry-stratified genome-wide association studies (GWASs), identifying an additional 5 sex-specific risk loci, 11 risk loci in European ancestry individuals and 1 risk locus in African ancestry individuals. We also performed a transcriptome-wide association study using expression quantitative trait loci from human aortic valves, discovering 54 new genes for which genetically predicted expression influences the risk of AS.
We then generated a new polygenic risk score for AS. Finally, we performed gene silencing experiments targeting biologically relevant genes identified by our GWAS. Silencing of CMKLR1 and LTBP4 in human valvular interstitial cells substantially decreased mineralization, implicating a role for polyunsaturated fatty acids and transforming growth factor β signaling in AS.
Reference: Aeron M. Small et al, Nature Genetics, 2025
An integrated view of the structure and function of the human 4D nucleome
The dynamic three-dimensional (3D) organization of the human genome (the 4D nucleome) is linked to genome function.
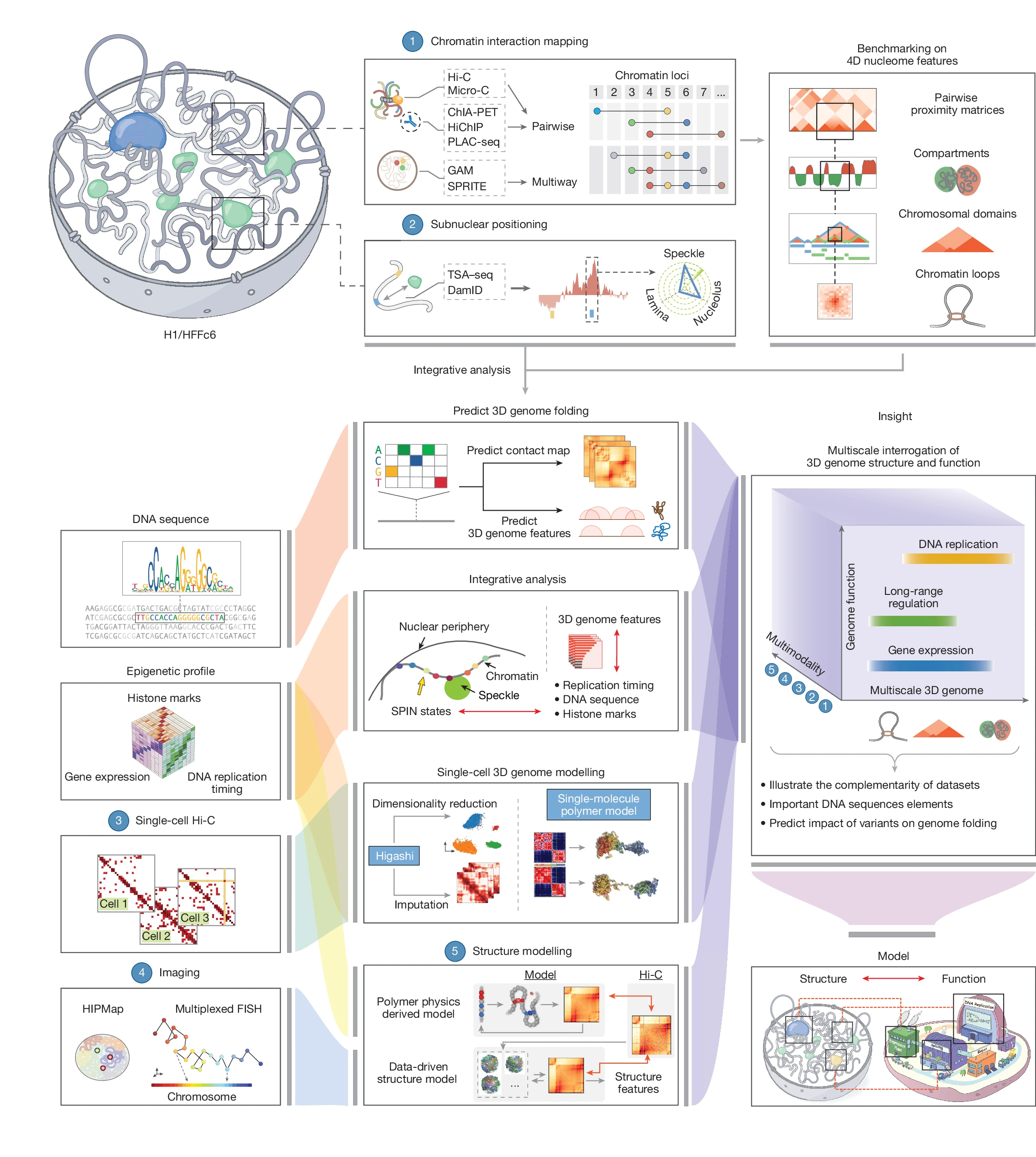
Here we describe efforts by the 4D Nucleome Project1 to map and analyse the 4D nucleome in widely used H1 human embryonic stem cells and immortalized fibroblasts (HFFc6). We produced and integrated diverse genomic datasets of the 4D nucleome, each contributing unique observations, which enabled us to assemble extensive catalogues of more than 140,000 looping interactions per cell type, to generate detailed classifications and annotations of chromosomal domain types and their subnuclear positions, and to obtain single-cell 3D models of the nuclear environment of all genes including their long-range interactions with distal elements.
Through extensive benchmarking, we describe the unique strengths of different genomic assays for studying the 4D nucleome, providing guidelines for future studies. Three-dimensional models of population-based and individual cell-to-cell variation in genome structure showed connections between chromosome folding, nuclear organization, chromatin looping, gene transcription and DNA replication. Finally, we demonstrate the use of computational methods to predict genome folding from DNA sequence, which will facilitate the discovery of potential effects of genetic variants, including variants associated with disease, on genome structure and function.
Reference: Job Dekker et al, Nature , 2025.
An open-source screening platform accelerates discovery of drug combinations
Drug combinations are essential to modern medicine, but their discovery remains slow and inefficient as experimental complexity expands rapidly with each additional drug tested. Although modern liquid handling systems enable complex and highly customizable experimental designs, a lack of strategies integrating these technologies with combination-specific analytical methods has limited throughput.
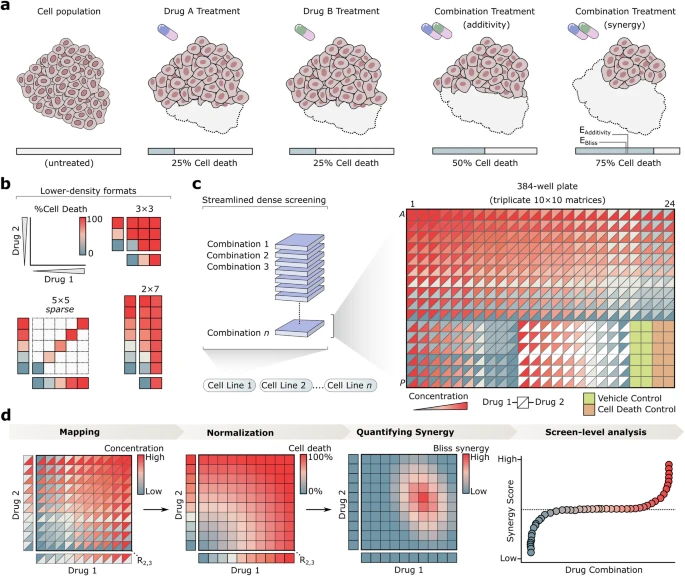
Here we introduce Combocat, an open-source and streamlined framework that combines acoustic liquid handling protocols with machine learning-based inference to achieve ultrahigh-throughput drug combination screening. Using Combocat, we generate a reference dataset of over 800 unique combinations in a dense 10 × 10 matrix format across multiple cell types, and use this to train a predictive model that accurately infers drug combination effects from sparse data, drastically reducing the number of experimental measurements required.
As proof of concept, we screened 9,045 combinations in a neuroblastoma cell line—the largest number of combinations tested in a single cell line to date—achieved using minimal resources. By integrating advanced drug dispensing technologies with predictive computational modeling, Combocat provides a scalable solution to accelerate the discovery of novel drug combinations.
Reference: William C. Wright et al, Nature Communications, 2025
Multimodal AI generates virtual population for tumor microenvironment modeling
The tumor immune microenvironment (TIME) critically impacts cancer progression and immunotherapy response. Multiplex immunofluorescence (mIF) is a powerful imaging modality for deciphering TIME, but its applicability is limited by high cost and low throughput.

We propose GigaTIME, a multimodal AI framework for population-scale TIME modeling by bridging cell morphology and states. GigaTIME learns a cross-modal translator to generate virtual mIF images from hematoxylin and eosin (H&E) slides by training on 40 million cells with paired H&E and mIF data across 21 proteins.
We applied GigaTIME to 14,256 patients from 51 hospitals and over 1,000 clinics across seven US states in Providence Health, generating 299,376 virtual mIF slides spanning 24 cancer types and 306 subtypes. This virtual population uncovered 1,234 statistically significant associations linking proteins, biomarkers, staging, and survival. Such analyses were previously infeasible due to the scarcity of mIF data. Independent validation on 10,200 TCGA patients further corroborated our findings.
Reference: Jeya Maria Jose Valanarasu et al, Cell, 2025
Whole-genome landscapes of 1,364 breast cancers
Breast cancer remains a major global health challenge. Here, to comprehensively characterize its genomic landscape and the clinical significance of genomic characteristics, we analysed whole-genome sequences from 1,364 clinically annotated breast cancers, with transcriptome data available for most cases.
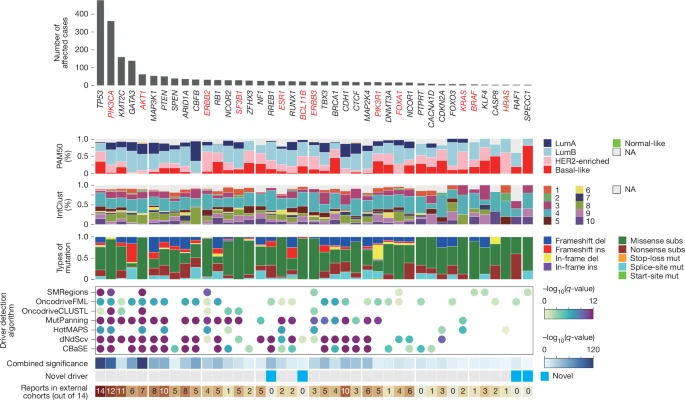
Our study expands the repertoire of oncogenic alterations and identifies novel driver genes, recurrent gene fusions, structural variants and copy number alterations. Timing analyses on copy number alterations suggest that genomic instability emerges decades before tumour diagnosis, and offer insights into early initiation of tumorigenesis.
Pattern-driven genomic features, including mutational signatures, homologous recombination deficiency, tumour mutational burden and tumour heterogeneity scores, were associated with clinical outcomes, highlighting their potential utility as predictive biomarkers for clinical evaluation of treatments such as CDK4/6 and HER2 inhibitors, as well as adjuvant and neoadjuvant chemotherapy. These findings highlight the power of large-scale, clinically annotated whole-genome sequencing in advancing our understanding of how genomic alterations shape patient outcomes.
Reference Ryul Kim, Jonghan Yu et al, Nature 2025.
A genome-wide association study of mass spectrometry proteomics using a nanoparticle enrichment platform
Most studies to date of protein quantitative trait loci (pQTLs) have relied on affinity proteomics platforms, which provide only limited information about the targeted protein isoforms and may be affected by genetic variation in their epitope binding.
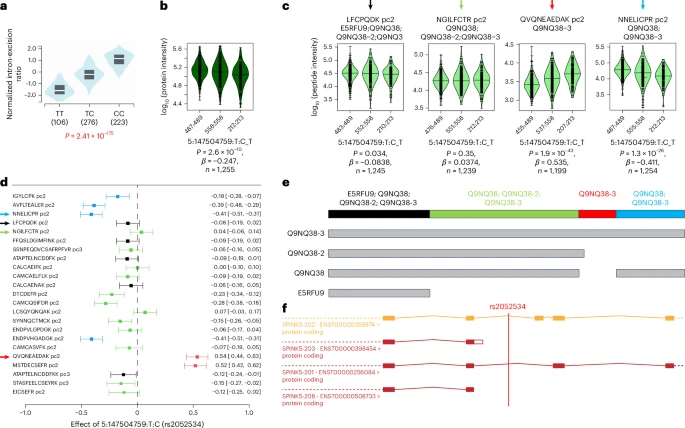
Here we show that mass spectrometry (MS)-based proteomics can complement these studies and provide insights into the role of specific protein isoform and epitope-altering variants. Using the Seer Proteograph nanoparticle enrichment MS platform, we identified and replicated new pQTLs in a genome-wide association study of proteins in blood plasma samples from two cohorts and evaluated previously reported pQTLs from affinity proteomics platforms.
We found that >30% of the evaluated pQTLs were confirmed by MS proteomics to be consistent with the hypothesis that genetic variants induce changes in protein abundance, whereas another 30% could not be replicated and are possibly due to epitope effects, although alternative explanations for nonreplication need to be considered on a case-by-case basis. Reference: Karsten Suhre et al, Nature Genetics 2025
Microbial signals in primary and metastatic brain tumors
Gliomas and brain metastases are associated with poor prognosis, necessitating a deeper understanding of brain tumor biology and the development of effective therapeutic strategies. Although our group and others have demonstrated microbial presence in various tumors, recent controversies regarding cancer-type-specific intratumoral microbiota emphasize the importance of rigorous, orthogonal validation.
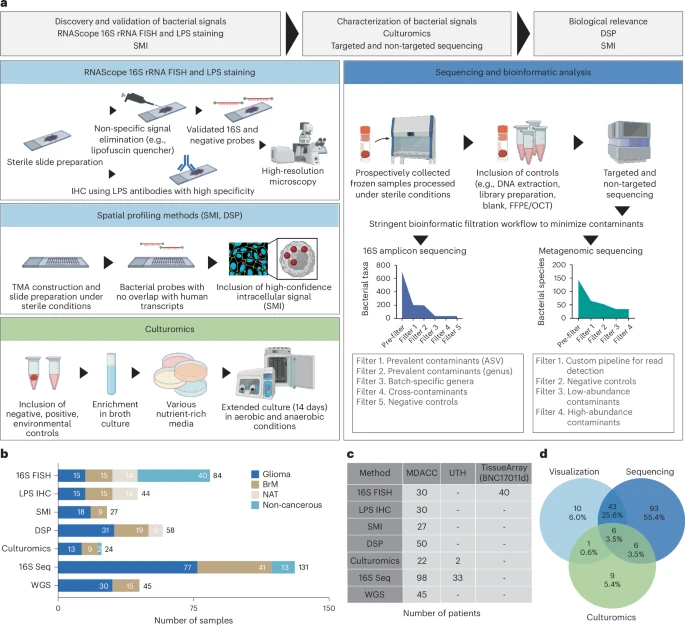
This prospective, multi-institutional study included a total of 243 samples from 221 patients, comprising 168 glioma and brain metastases samples and 75 non-cancerous or tumor-adjacent tissues. Using stringent fluorescence in situ hybridization, immunohistochemistry and high-resolution spatial imaging, we detected intracellular bacterial 16S rRNA and lipopolysaccharides in both glioma and brain metastases samples, localized to tumor, immune and stromal cells.
Custom 16S and metagenomic sequencing workflows identified taxa associated with intratumoral bacterial signals in the tumor microenvironment; however, standard culture methods did not yield readily cultivable microbiota. Spatial analyses revealed significant correlations between bacterial 16S signals and antimicrobial and immunometabolic signatures at regional, neighborhood and cellular levels. Furthermore, intratumoral 16S bacterial signals showed sequence overlap with matched oral and gut microbiota, suggesting a possible connection with distant communities. Together, these findings introduce microbial elements as a component of the brain tumor microenvironment and lay the foundation for future mechanistic and translational studies. Reference: Golnaz Morad et al, Nature Medicine, 2025
GWAS identify distinct genetic architectures for early-onset and late-onset depression
Major depressive disorder (MDD) is a common and heterogeneous disorder of complex etiology. Studying more homogeneous groups stratified according to clinical characteristics, such as age of onset, can improve the identification of the underlying genetic causes and lead to more targeted treatment strategies.
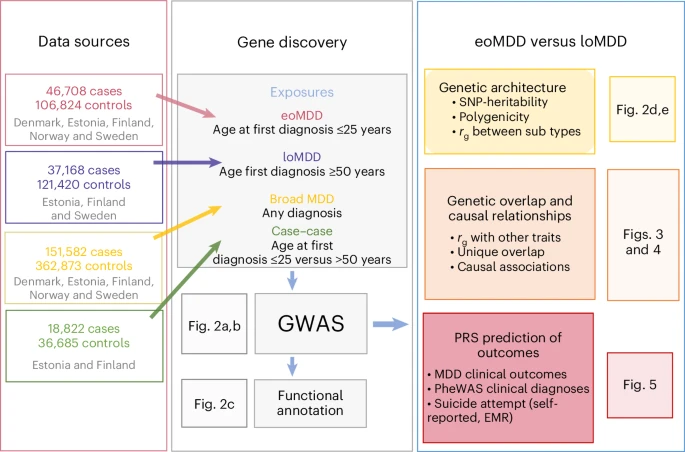
We leveraged Nordic biobanks with longitudinal health registries to investigate differences in the genetic architectures of early-onset (eoMDD; n = 46,708 cases) and late-onset (loMDD; n = 37,168 cases) MDD. We identified 12 genomic loci for eoMDD and two for loMDD. Overall, the two MDD subtypes correlated moderately (genetic correlation, rg = 0.58) and differed in their genetic correlations with related traits.
These findings suggest that eoMDD and loMDD have partially distinct genetic signatures, with a specific developmental brain signature for eoMDD. Importantly, we demonstrate that polygenic risk scores (PRS) for eoMDD predict suicide attempts within the first 10 years after the initial diagnosis: the absolute risk for suicide attempt was 26% in the top PRS decile, compared to 12% and 20% in the bottom decile and the intermediate group, respectively. Taken together, our findings can inform precision psychiatry approaches for MDD. Reference: John R. Shorter et al, Nature Genetics (2025)